Alberto Ferlin
Università di Padova, Dipartimento di Medicina, UOC Andrologia e Medicina della Riproduzione
DEFINIZIONE E CLASSIFICAZIONE
Per criptorchidismo si intende l’assenza di uno o entrambi i testicoli nella borsa scrotale alla nascita, con arresto lungo il fisiologico tragitto di discesa dall’addome. Sebbene il criptorchidismo sia spesso considerato una patologia di lieve entità, in realtà esso rappresenta l’anomalia congenita più frequente dell’apparato urogenitale ed è il più importante fattore di rischio per infertilità e tumore testicolare in età adulta. Una diagnosi precoce e una corretta gestione del testicolo criptorchide sono pertanto necessari, soprattutto per preservare la fertilità, eseguire un corretto counselling e follow-up del paziente e per ridurre il rischio di trasformazione neoplastica dei testicoli e l’eventuale insorgenza di ipogonadismo.
Il criptorchidismo è bilaterale in un terzo dei casi e monolaterale nei due terzi. I testicoli criptorchidi vengono classificati in base alla loro posizione lungo il tragitto di discesa (sede addominale alta/bassa, inguinale, sovra-scrotale, alto-scrotale) e vengono quindi distinti dai testicoli ectopici, che sono localizzati al di fuori della fisiologica via di discesa. Tuttavia, nella pratica clinica e per indirizzare la terapia risulta utile anche una semplice distinzione tra testicoli palpabili e non palpabili e tra forme bilaterali e monolaterali. Una condizione particolare è rappresentata dall’assenza di uno o entrambi i testicoli, condizione nota come anorchia o sindrome del testicolo evanescente.
Il criptorchidismo può essere un’anomalia isolata o più raramente si può associare ad altre malformazioni dell’apparato uro-genitale o può far parte di sindromi genetiche più complesse. Il criptorchidismo va anche distinto dal testicolo retrattile (testicolo normalmente disceso alla nascita, che risale in canale inguinale e può essere riportato manualmente in sede scrotale, da dove risale per riflesso cremasterico), dal criptorchidismo acquisito (testicolo normalmente disceso alla nascita e poi risalito in canale inguinale, da dove non è più riposizionabile manualmente nello scroto) e dal testicolo mobile (testicolo non criptorchide alla nascita, che si muove facilmente per effetto del muscolo cremastere fuori dal sacco scrotale, ma vi ritorna altrettanto facilmente).
| Tabella 1 Classificazione dei difetti di posizione testicolare |
|
| Testicolo in sede | In posizione scrotale, che non risale mai nel canale inguinale, nemmeno per riflesso cremasterico o con manipolazione |
| Criptorchidismo congenito | Assenza di uno o entrambi i testicoli nello scroto, per arresto lungo il fisiologico tragitto di discesa testicolare |
| Testicolo in ascensore (ascending testis o criptorchidismo acquisito) | Testicolo non criptorchide alla nascita o testicolo criptorchide disceso spontaneamente e poi risalito stabilmente in canale inguinale (difficile manipolazione) |
| Gliding testis | Appena sotto l’anello inguinale esterno e posizionabile manualmente solo in sede alto-scrotale, da dove risale immediatamente |
| Testicolo retrattile o alto-scrotale | In posizione scrotale alta, posizionabile manualmente in sacca scrotale, dove può rimanere per un certo periodo, e retrattile in sede alta per riflesso cremasterico |
| Testicolo mobile o migrante | In posizione scrotale, risale in canale inguinale spontaneamente (riflesso cremasterico, rapporti sessuali, freddo, ecc) o con manipolazione e ritorna in sacca scrotale spontaneamente o con manipolazione |
| Testicolo ectopico | Assenza di uno o entrambi i testicoli nello scroto, con localizzazione al di fuori del fisiologico tragitto di discesa testicolare |
CENNI DI EMBRIOLOGIA
La discesa del testicolo durante la vita fetale, dalla posizione originaria in prossimità del rene fino alla borsa scrotale, è un meccanismo complesso che richiede l’interazione di fattori anatomici, meccanici e ormonali. Si distinguono due fasi principali:
- trans-addominale (tra la 10° e la 23° settimana gestazionale), che porta il testicolo in prossimità dell’orifizio inguinale interno;
- inguino-scrotale (dalla 26°-28° settimana fino alla nascita), che porta il testicolo nella sua posizione definitiva nella borsa scrotale omolaterale.
La discesa del testicolo è regolata da due ormoni principali, prodotti dalle cellule di Leydig: Il testosterone, oltre a essere il principale ormone per lo sviluppo sessuale in senso maschile del feto e per il corretto sviluppo degli organi genitali esterni, è il maggior attore della fase inguino-scrotale della discesa testicolare; l’INSL3 è il maggior responsabile della fase trans-addominale. È di ausilio in questo processo l’ormone anti-mulleriano (AMH) prodotto dalle cellule di Sertoli, che provoca la regressione dei dotti di Müller.
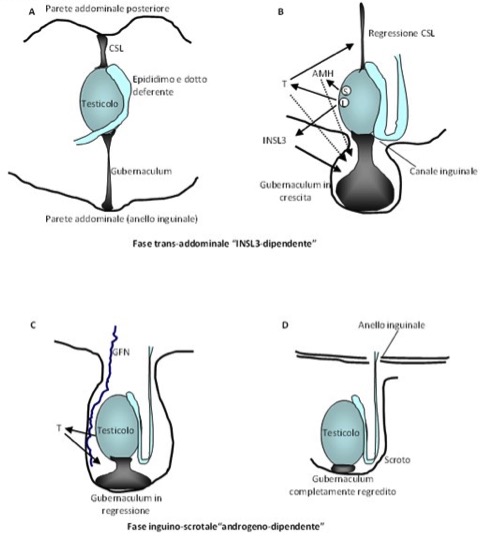
Figura 1. Schema della discesa testicolare.
(A) La differenziazione della gonade indifferenziata in presenza del cromosoma Y porta alla produzione di AMH dalle cellule di Sertoli (S) e di testosterone e INSL3 dalle cellule di Leydig
(B) L’effetto diretto e indiretto (attraverso il nervo genito-femorale – GNF – e il CGRP) di questi 2 ormoni, principalmente sul legamento sospensore craniale (CSL) e sul gubernaculum, è alla base del processo bifasico di discesa testicolare.
(C) La regressione del CSL è soprattutto sotto il controllo del testosterone, mentre INSL3 agisce principalmente sul gubernaculum, sul quale agiscono in misura minore AMH e testosterone, verosimilmente attraverso il GNF.
EPIDEMIOLOGIA
Il criptorchidismo interessa circa il 3-5% dei bambini nati a termine e il 9-30% dei pre-termine. Anche il basso peso alla nascita è un importante fattore di rischio: la prevalenza nei nati con peso < 2.5 kg è di circa il 20-25%. L’incidenza del criptorchidismo sembra essere aumentata negli ultimi decenni, soprattutto in alcuni paesi, probabilmente come conseguenza dell’esposizione a fattori ambientali con attività simil-ormonale, soprattutto di tipo estrogenico e/o anti-androgenico.
Circa la metà dei testicoli criptorchidi alla nascita discende spontaneamente entro i 4-6 mesi di vita, soprattutto nei nati pre-termine, e pertanto la prevalenza del criptorchidismo a un anno di vita è circa l’1-2%. La terapia dei testicoli criptorchidi non dovrebbe pertanto iniziare prima del 4°-6° mese.
EZIOPATOGENESI
Le cause del criptorchidismo sono molteplici (tab 1), ma nella maggior parte dei casi non si riscontrano fattori eziologici certi. I fattori di rischio più importanti sono rappresentati dalla prematurità e dal basso peso alla nascita, ma sembrano avere un ruolo anche il diabete in gravidanza e il fumo.
Il criptorchidismo può anche far parte del corteo sindromico di molte malattie genetiche complesse, la cui incidenza è comunque molto bassa.
| Tabella 2 Cause principali di criptorchidismo |
|||
| Cause | Tipi | Commenti | |
| Idiopatiche | Fattori di rischio maggiori: basso peso alla nascita, prematurità, piccolo per età gestazionale (SGA) | ||
| Endocrine | Ipogonadismo ipogonadotropo (idiopatico, sindrome di Kallmann, altri difetti genetici) Ipogonadismo ipergonadotropo |
Non sono frequenti, ma alcuni autori sostengono che tutti i bambini criptorchidi abbiano un certo grado di ipogonadismo relativo | |
| Genetiche | Monogeniche | Mutazioni del gene INSL3 e del suo recettore (RXFP2) Mutazioni del gene per il recettore degli androgeni |
Circa il 5-10% dei casi non associati ad altre anomalie dell’apparato uro-genitale; più frequenti nei casi di criptorchidismo bilaterale, non si associano a discesa spontanea dei testicoli dopo la nascita |
| Cromosomiche e sindromi genetiche complesse | S. di Klinefelter S. del maschio 46,XX S. da insensibilità periferica agli androgeni S. di Down S. di Noonan S. di Beckwith-Wiedemann S. di Prader-Willi FG syndrome (o s. di Opitz-Kaveggia) Del 22q11.2 Del 1p36 |
||
| Fattori materni | Diabete Fumo Alcool Assunzione in gravidanza di sostanze ad attività estrogenica o anti-androgenica |
||
| Anomalie anatomiche | Impervietà del canale inguinale Ernia inguinale Brevità del funicolo spermatico |
Frequenti | |
DIAGNOSI
La diagnosi è volta soprattutto a differenziare il criptorchidismo dalle altre anomalie di discesa e posizione del testicolo, soprattutto dall’anorchia e dall’ectopia.
L’anamnesi e l’esame obiettivo possono abbastanza facilmente individuare il testicolo mobile, testicolo che discende spontaneamente nello scroto e risale nel canale inguinale per effetto della trazione del muscolo cremastere. Nella maggior parte di questi casi non è necessaria nessuna terapia, ma il bambino potrà essere controllato annualmente per assicurarsi che la situazione si regolarizzi spontaneamente e in maniera definitiva all’epoca dello sviluppo puberale.
L’esame obiettivo è indirizzato all’individuazione della posizione del testicolo, del suo volume e consistenza e della sua mobilità verso il sacco scrotale. Quando non si riesce a palpare il testicolo e quindi bisogna differenziare tra criptorchidismo, ectopia e anorchia congenita, sono necessari ulteriori accertamenti. L’anorchia congenita bilaterale può essere sospettata per la frequente coesistenza di micropene e di marcata ipoplasia dello scroto e va poi confermata con un test con hCG, che documenta un’assente risposta del testosterone.
Utile come esame di primo livello è anche l’ecografia testicolare e solo quando il testicolo non è dimostrabile nemmeno con tale indagine, si può ricorrere alla RMN o, meglio ancora, alla laparoscopia o esplorazione chirurgica. L’esplorazione chirurgica è di regola determinante per distinguere fra l’agenesia di un solo testicolo e un criptorchidismo monolaterale completo.
Solamente in alcuni casi (criptorchidismo bilaterale con testicoli non palpabili, associato o meno a ipospadia, micropene o genitali ambigui) è necessario un approfondimento endocrinologico e genetico, che include cariotipo, dosaggi ormonali (testosterone, LH, FSH, 17-OH-progesterone, o altre indagini ormonali secondo l’indicazione clinica); i test al GnRH e all’hCG sono necessari solo in una minoranza di casi.
L’analisi genetica del gene per il recettore degli androgeni, il gene INSL3 ed il suo recettore (RXFP2) possono essere utili anche nei casi di criptorchidismo monolaterale. In casi selezionati potranno essere utili altre analisi genetiche in base al sospetto clinico (per esempio analisi dei geni dell’ipogonadismo ipogonadotropo).
TERAPIA
La correzione del criptorchidismo ha lo scopo di riportare i testicoli nella loro sede fisiologica, soprattutto per prevenire (o quantomeno ridurre) la degenerazione della funzione testicolare e l’insorgenza di tumori del testicolo o quantomeno renderne più agevole la diagnosi precoce. Inoltre, il trattamento del testicolo criptorchide ha anche risvolti psicologici legati alla presenza di uno scroto vuoto.
La correzione del criptorchidismo è attualmente raccomandata entro il secondo anno di vita, meglio se entro l’anno e mezzo. La terapia ideale del criptorchidismo è chirurgica (orchidopessi), essendo ormai la terapia farmacologica (hCG in associazione o meno con hMG, GnRH in associazione o meno con hMG) considerata priva di evidenze di efficacia e ad alto rischio di recidiva. L’intervento chirurgico ha successo definitivo in più del 95% dei casi, con una bassa incidenza (1%) di complicanze.
FOLLOW-UP
Rischio di infertilità
Uomini con storia di criptorchidismo sono frequentemente infertili e una storia di criptorchidismo si riscontra in circa il 10% dei soggetti infertili e nel 20% di quelli azoospermici. Generalmente i danni alla spermatogenesi sono più gravi nelle forme di criptorchidismo bilaterale e addominale. Tuttavia, anche nei casi di criptorchidismo monolaterale ci può essere un danno spermatogenetico nel testicolo controlaterale. In media, circa il 70-80% dei soggetti con criptorchidismo bilaterale e il 40-50% dei soggetti con criptorchidismo monolaterale presenta oligozoospermia o azoospermia.
L’età all’orchidopessi ha influenza sul successivo deterioramento della spermatogenesi. L’orchidopessi precoce però, seppur in grado di ridurre il rischio di infertilità, non lo annulla.
Secondo queste considerazioni è utile informare i soggetti con criptorchidismo (e i familiari) circa il rischio di infertilità in età adulta ed eseguire controlli periodici del liquido seminale dai 16-18 anni, con eventuale crio-conservazione dello stesso prima che il danno spermatogenetico progressivo sia definitivo.
Rischio di tumore testicolare
Il criptorchidismo rappresenta uno dei principali fattori di rischio per il tumore testicolare, essendo il rischio relativo 4-8 volte superiore rispetto alla popolazione generale e maggiore per i casi di criptorchidismo bilaterale e per i casi di criptorchidismo addominale. Circa il 5-10% dei casi di tumore testicolare (tumori a cellule germinali) insorge in soggetti ex-criptorchidi. Nei casi di criptorchidismo monolaterale, si può sviluppare tumore anche nel testicolo controlaterale (nel 15-20% dei casi).
Pertanto, è opportuno eseguire un follow-up dei soggetti con storia di criptorchidismo, tenendo presente che il tumore del testicolo ha un picco di incidenza tra i 15 e i 40 anni e il rischio di tumore anche nel testicolo controlaterale nei casi di criptorchidismo monolaterale. In tal senso può essere utile informare i ragazzi ex-criptorchidi (e i familiari) circa l’utilità dell’auto-palpazione dei testicoli ed eseguire controlli clinici periodici con cadenza annuale (visita, ecografia testicolare).
Rischio di ipogonadismo
Sia nei casi di criptorchidismo bilaterale (più frequentemente) che monolaterale (più raramente) esiste spesso un danno testicolare bilaterale, progressivo nel tempo, non completamente influenzato anche da un’orchidopessi precoce, che coinvolge sia la componente spermatogenetica che endocrina. I soggetti con criptorchidismo pertanto, indipendentemente dalla sua gravità e timing di correzione, dovrebbero essere considerati soggetti ad alto rischio di ipogonadismo, opportunamente informati di conseguenza e seguiti nel tempo, dallo sviluppo puberale (possibilità di ritardo puberale, ginecomastia) in poi, con monitoraggio della crescita testicolare e dosaggi ormonali.
| Tabella 3 Management e follow-up del criptorchidismo |
||
| Età | Cosa fare |
Persone coinvolte |
| Nascita | Esaminare la posizione dei testicoli, distinguere le diverse forme di anomalie di discesa e posizione, valutare malformazioni associate per possibile DSD Notare età gestazionale, peso alla nascita, SGA Possibili fattori materni (diabete, disruptors/farmaci) Counselling ai genitori |
Pediatra/endocrinologo pediatrico Chirurgo/urologo pediatrico Genetista Genitori |
| 6-12 mesi | Verificare discesa spontanea ed eventuale criptorchidismo acquisito Considerare dosaggio FSH, LH, testosterone, AMH, test con hCG nel caso di testicoli non palpabili Considerare cariotipo, analisi genetica INSL3, RXFP2, AR, geni per ipogonadismo ipogonadotropo o altre analisi genetiche selezionate Considerare esplorazione chirurgica Programmare orchidopessi entro i 2 anni Counselling ai genitori |
Pediatra/endocrinologo pediatrico Chirurgo/urologo pediatrico Genetista Pediatra di libera scelta Genitori |
| Pre-pubertà | Esame obiettivo dei testicoli a ogni visita pediatrica Counselling ai genitori |
Pediatra di libera scelta Genitori |
| Pubertà | Monitorare sviluppo puberale e testicolare + eventuali approfondimenti specifici Counselling al paziente e genitori |
Pediatra/endocrinologo pediatrico Pediatra di libera scelta Paziente e genitori |
| 16-18 anni | Transizione all’endocrinologo dell’adulto/andrologo per valutazione della funzione gonadica (esame del liquido seminale + ev. crio-conservazione e approfondimenti specifici; ecografia testicolare; FSH, LH, testosterone; ecc) Counselling al paziente e genitori: auto-palpazione, rischio di infertilità, tumori e ipogonadismo |
Endocrinologo dell’adulto/andrologo Paziente e genitori |
| > 18 anni | Esame obiettivo dei testicoli alle visite MMG e sportive Presa in carico da parte dell’endocrinologo dell’adulto/andrologo per follow-up annuale |
MMG, medici sportivi Endocrinologo dell’adulto/andrologo Paziente |
BIBLIOGRAFIA
- Abaci A, et al. Epidemiology, classification and management of undescended testes: does medication have value in its treatment? J Clin Res Pediatr Endocrinol 2013, 5: 65-72.
- Bay K, et al. Testicular descent: INSL3, testosterone, genes and the intrauterine milieu. Nat Rev Urol 2011, 8: 187-96.
- Ferlin A, et al. Genetic alterations associated with cryptorchidism. JAMA 2008, 300: 2271-6.
- Foresta C, et al. Role of hormones, genes, and environment in human cryptorchidism. Endocr Rev 2008, 29: 560-80.
- Hutson JM, et al. Cryptorchidism. Semin Pediatr Surg 2010, 19: 215-24.
- Kolon TF, et al. Evaluation and treatment of cryptorchidism: AUA guideline. J Urol 2014, 192: 337-45.
- Lee PA, et al. Cryptorchidism. Curr Opin Endocrinol Diabetes Obes 2013, 20: 210-6.
- Ritzén EM. Undescended testes: a consensus on management. Eur J Endocrinol 2008, 159 suppl 1: S87-90.
